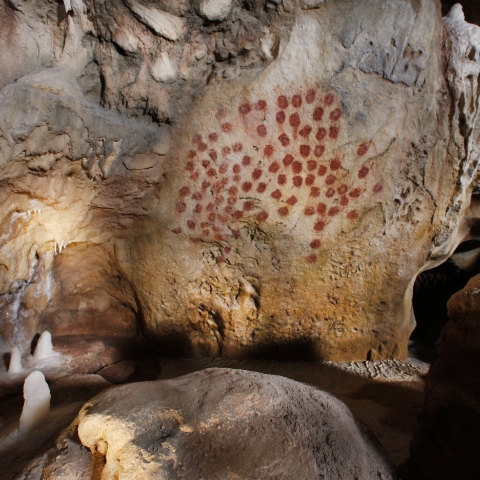
Inscrite au patrimoine mondial de l’Unesco, la grotte Chauvet rassemble des dessins d’une qualité exceptionnelle datés d’il y a 36 000 ans. Découvrez son espace de restitution !
La Grotte Chauvet 2
Une expérience unique au coeur d’un parc de 29 hectares